 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInterpretable Machine Learning for Quantum-Informed Property Predictions in Artificial Sensing Materials
Jan 01, 2026Digital sensing faces challenges in developing sustainable methods to extend the applicability of customized e-noses to complex body odor volatilome (BOV). To address this challenge, we developed MORE-ML, a computational framework that integrates quantum-mechanical (QM) property data of e-nose molecular building blocks with machine learning (ML) methods to predict sensing-relevant properties. Within this framework, we expanded our previous dataset, MORE-Q, to MORE-QX by sampling a larger conformational space of interactions between BOV molecules and mucin-derived receptors. This dataset provides extensive electronic binding features (BFs) computed upon BOV adsorption. Analysis of MORE-QX property space revealed weak correlations between QM properties of building blocks and resulting BFs. Leveraging this observation, we defined electronic descriptors of building blocks as inputs for tree-based ML models to predict BFs. Benchmarking showed CatBoost models outperform alternatives, especially in transferability to unseen compounds. Explainable AI methods further highlighted which QM properties most influence BF predictions. Collectively, MORE-ML combines QM insights with ML to provide mechanistic understanding and rational design principles for molecular receptors in BOV sensing. This approach establishes a foundation for advancing artificial sensing materials capable of analyzing complex odor mixtures, bridging the gap between molecular-level computations and practical e-nose applications.
Accurate Machine Learned Quantum-Mechanical Force Fields for Biomolecular Simulations
May 17, 2022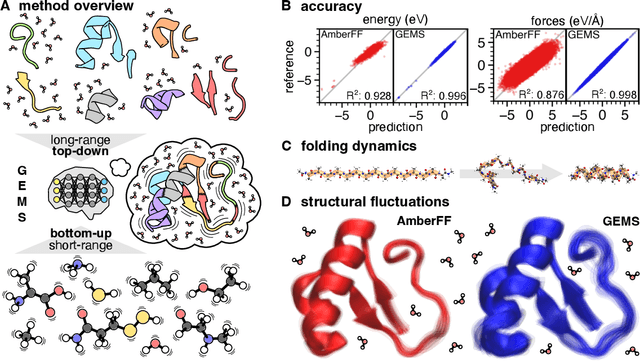
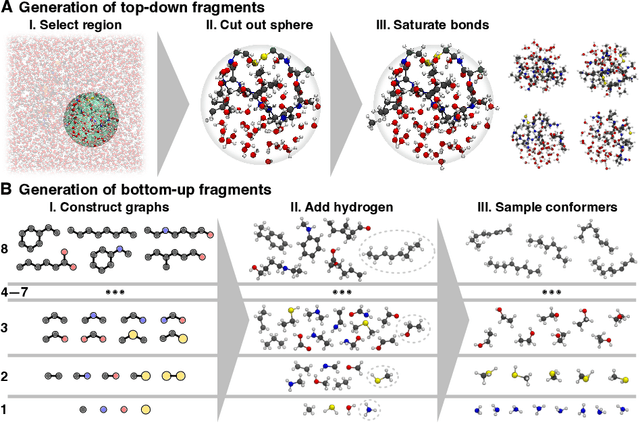
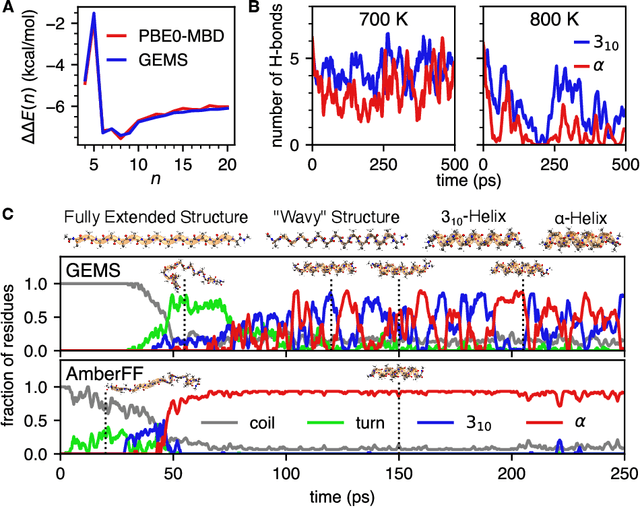
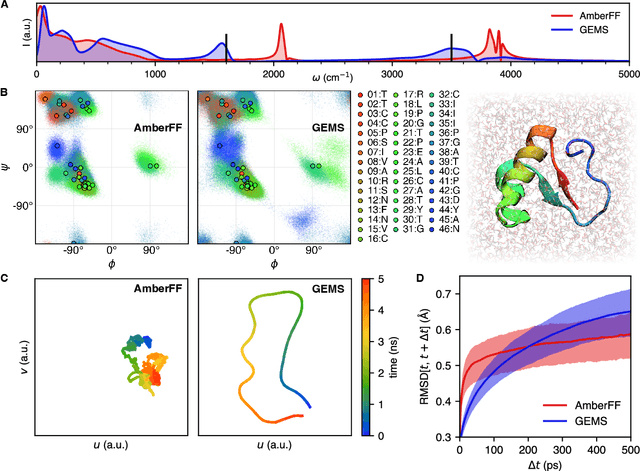
Molecular dynamics (MD) simulations allow atomistic insights into chemical and biological processes. Accurate MD simulations require computationally demanding quantum-mechanical calculations, being practically limited to short timescales and few atoms. For larger systems, efficient, but much less reliable empirical force fields are used. Recently, machine learned force fields (MLFFs) emerged as an alternative means to execute MD simulations, offering similar accuracy as ab initio methods at orders-of-magnitude speedup. Until now, MLFFs mainly capture short-range interactions in small molecules or periodic materials, due to the increased complexity of constructing models and obtaining reliable reference data for large molecules, where long-ranged many-body effects become important. This work proposes a general approach to constructing accurate MLFFs for large-scale molecular simulations (GEMS) by training on "bottom-up" and "top-down" molecular fragments of varying size, from which the relevant physicochemical interactions can be learned. GEMS is applied to study the dynamics of alanine-based peptides and the 46-residue protein crambin in aqueous solution, allowing nanosecond-scale MD simulations of >25k atoms at essentially ab initio quality. Our findings suggest that structural motifs in peptides and proteins are more flexible than previously thought, indicating that simulations at ab initio accuracy might be necessary to understand dynamic biomolecular processes such as protein (mis)folding, drug-protein binding, or allosteric regulation.